Ground State Solving
The Variational Quantum Eigensolver (VQE) is a well-known algorithm for its applications in the chemistry field. The Classiq platform uses this algorithm to solve ground state problems.
Background
Calculating the energies of a molecular Hamiltonian, especially the ground state, allows insights into the chemical properties of the molecule. Since the molecular Hamiltonian is inherently quantum, quantum computing calculates such properties efficiently by leveraging the same quantum effects in appropriate quantum algorithms.
Steps:
- Define the molecule and calculate its Hamiltonian operator.
- Construct an appropriate variational quantum model, containing the description of the quantum program (the ansatz) and the classical flow (VQE). Then synthesize the model to a quantum program.
- Optimize the parameters in the ansatz by executing the hybrid quantum-classical algorithm to find the Hamiltonian (or molecule) ground state.
The process is described visually in the diagram:
flowchart LR
classDef default fill:#FFFFFF,stroke:#000000,stroke-width:1px;
A(Define Molecule and\nGenerate Hamiltonian) --> B(Define an\nAnsatz);
B --> C(Optimize its\nparameters);Defining a Molecule and Calculating Its Hamiltonian
Given a molecule, what is the full Hamiltonian describing the interactions between all electrons and nuclei?
Following is an example of calculating the Hamiltonian of the hydrogen molecule.
In the IDE Model page - select the Chemistry option from the Built In Apps folder in the Models pane:
Enter the following molecule model in the Chemistry problem editor:
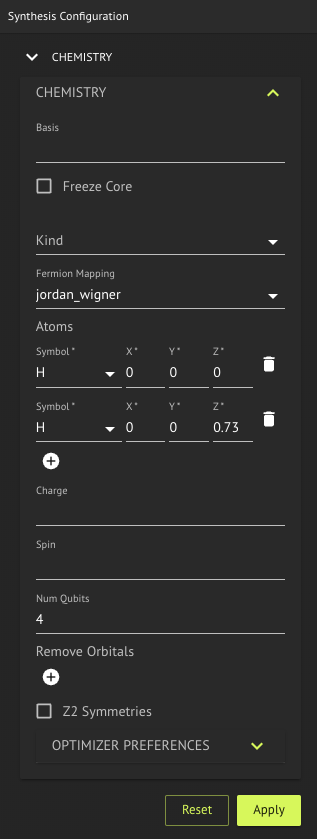
If you edit the molecule model make sure to click the Apply button. You should now see the Hamiltonian in compact format in the Model Editor.
from classiq.applications.chemistry import Molecule, MoleculeProblem
molecule = Molecule(
atoms=[
("H", (0.0, 0.0, 0.0)),
("H", (0.0, 0.0, 0.735)),
],
)
gs_problem = MoleculeProblem(
molecule=molecule,
mapping="jordan_wigner",
)
hamiltonian = gs_problem.generate_hamiltonian()
The example above defines a hydrogen molecule by specifying its two hydrogen
atoms and their position in a 3D space.
The molecule is then used to create a MoleculeProblem, together with the following optional parameters:
mapping: str– The mapping between the fermionic Hamiltonian and a Pauli string Hamiltonian that can be applied on the qubits. Supported types:"jordan_wigner""parity""bravyi_kitaev""fast_bravyi_kitaev"freeze_core: bool– Remove the "core" orbitals of the atoms defining the molecule.two_qubit_reduction: bool– Perform two qubit reduction if possible.-
z2_symmetries: bool– Perform Z2 symmetries reduction [1] . -
Currently, the only supported basis set is
"sto3g"(see PySCF).
Finally, the Hamiltonian can be generated from the MoleculeProblem, if needed.
Constructing a Variational Quantum Model
The ground state of a Hamiltonian is found using an appropriate variational quantum program (usually called an ansatz), which is combined with a classical optimizer to find the minimal expectation value for the Hamiltonian. See the list of built-in ansatzes.
Unitary Coupled Cluster Ansatz Example
This example shows the generation of the commonly used, chemistry-inspired UCC ansatz, which is a unitary version of the classical coupled cluster (CC) method [2] . First, initialize the quantum program to the Hartree-Fock state, then apply the UCC function. See details of the UCC ansatz in UCC.
After generating a Hamiltonian as described above, you can either Synthesize the quantum program or Re-edit the the chemistry model and the Hamiltonain.
Synthesistakes you to the Quantum Program tab.
from classiq import construct_chemistry_model, synthesize, show
from classiq.applications.chemistry import Molecule, MoleculeProblem
from classiq.applications.chemistry import UCCParameters, ChemistryExecutionParameters
from classiq.execution import OptimizerType
molecule = Molecule(
atoms=[("H", (0.0, 0.0, 0.0)), ("H", (0.0, 0.0, 0.735))],
)
gs_problem = MoleculeProblem(
molecule=molecule,
mapping="jordan_wigner",
)
ucc_params = UCCParameters(excitations=[1, 2])
execution_params = ChemistryExecutionParameters(
optimizer=OptimizerType.COBYLA,
max_iteration=30,
)
# The construct_chemistry_model function wraps the quantum and classical parts together, and creates a full hybrid model.
model = construct_chemistry_model(
chemistry_problem=gs_problem,
use_hartree_fock=True,
ansatz_parameters=ucc_params,
execution_parameters=execution_params,
)
qprog = synthesize(model)
show(qprog)
This is the output quantum program.
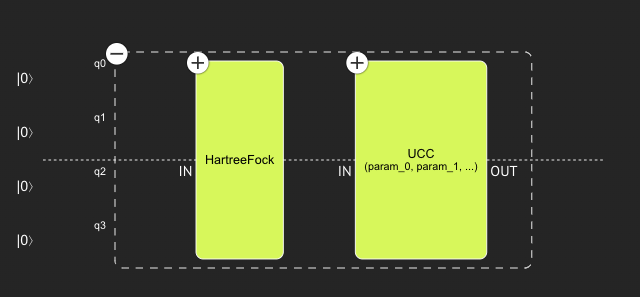 .
.
Optimizing to Find the Hamiltonian Ground State
After you specify a molecular Hamiltonian and an ansatz to use in the algorithm, combine everything and execute the VQE algorithm to find the Hamiltonian ground state.
Ensure you are in the Execution tab.
Click the Run button in the same way that you execute other quantum programs throughout the platform.
In this case, the Classiq engine executes the hybrid algorithm, optimizing the parameters in the ansatz
to find the minimum of the objective function \(\left\langle \psi(\theta) \middle| H \middle| \psi(\theta) \right\rangle\).
This process attempts to find the molecule's ground state.
from classiq import construct_chemistry_model, synthesize, show
from classiq.applications.chemistry import Molecule, MoleculeProblem
from classiq.applications.chemistry import UCCParameters, ChemistryExecutionParameters
from classiq.execution import OptimizerType
from classiq import execute
molecule = Molecule(
atoms=[("H", (0.0, 0.0, 0.0)), ("H", (0.0, 0.0, 0.735))],
)
gs_problem = MoleculeProblem(
molecule=molecule,
mapping="jordan_wigner",
)
ucc_params = UCCParameters(excitations=[1, 2])
execution_params = ChemistryExecutionParameters(
optimizer=OptimizerType.COBYLA,
max_iteration=30,
)
# The construct_chemistry_model function wraps the quantum and classical parts together, and creates a full hybrid model.
model = construct_chemistry_model(
chemistry_problem=gs_problem,
use_hartree_fock=True,
ansatz_parameters=ucc_params,
execution_parameters=execution_params,
)
qprog = synthesize(model)
result = execute(qprog).result()
execute(qprog) executes the hybrid algorithm, optimizing the parameters in the ansatz
to find the minimum of the objective function \(\left\langle \psi(\theta) \middle| H \middle| \psi(\theta) \right\rangle\).
This process attempts to find the molecule's ground state.
Compare the VQE result to the exact result for small molecules obtained by calculating the ground state exact solution.
For details of the resulting object (for molecule problems), see GroundStateResult.
Additional Examples
See further ansatz examples: hardware efficient ansatz, Hamiltonian variational ansatz.
Troubleshooting the IDE
- If the "Execute for Hamiltonian" checkbox is missing:
- The checkbox only appears when there is a Hamiltonian available for the quantum program. It might be missing if:
- You clicked the
Synthesizebutton before theGenerate Hamiltonianbutton. - You used the SDK to synthesize the quantum program, but the IDE to execute it. Classiq does not support Hamiltonians generated in the SDK.
- You clicked the
References
[1] Bravyi, Sergey, Jay M. Gambetta, Antonio Mezzacapo, and Kristan Temme, Tapering off qubits to simulate fermionic Hamiltonians (2017).
[2] Panagiotis K. Barkoutsos, Jerome F. Gonthier, Igor Sokolov, Nikolaj Moll, Gian Salis, Andreas Fuhrer, Marc Ganzhorn, Daniel J. Egger, Matthias Troyer, Antonio Mezzacapo, Stefan Filipp, and Ivano Tavernelli, Quantum algorithms for electronic structure calculations: Particle-hole Hamiltonian and optimized wave-function expansions Phys. Rev. A 98, 022322 (2018).