

View on GitHub
Open this notebook in GitHub to run it yourself
The FermionHamiltonianProblem class: Defining an Electronic structure problem.
There are two ways of defining an electronic structure problem, either providing MolecularData of a molecule, or by directly defining a Fermionic Hamiltonian together with the number of spin up/down () particles.
Below we demonstrate the former, which is a more practical usecase.
For the direct definition see this example.
Defining a molecule
We start with defining a molecule, specifying its geometry (elements and their 3D position), multiplicity (), basis, and an optional string for its description. In this tutorial we focus on the LiH molecule. There are several ways to get geometry of molecules, typical way involves using the SMILES (Simplified Molecular Input Line Entry System) of a molecule and use a chemical package such asRDkit to extract the geometry as an xyz file (a code example is given in the Appendix A of this notebook).
For simplicity, we store the geometry in advance in the lih.xyz file and load it.
Comment: For complex molecules it is possible to call directly from openfermion.chem import geometry_from_pubchem
Output:
save and load methods.
Output:
Defining a reduced problem (active space/freeze core)
In some cases, we can “freeze” some of the orbitals, occupying them with both spin up and spin down electrons. This will be orbitals with very low energy, such as the core orbitals, which are expected to be “classically” (with both spin up and down) occupied. In addition, we can exclude orbitals with very high energies, as they are unlikely to contribute significantly to the ground state. In other words, we can choose the active space for our molecular problem --- the spatial orbitals that are relevant to the quantum problem. This of-course reduces the problem we need to tackle. Below we define aFermionHamiltonianProblem for the LiH molecule, freezing its core () orbital.
The updated number of spatial orbitals and electrons are a property of the class.
Output:
Output:
Orbital labeling: For spatial orbitals we have electron orbitals. The Classiq object for the electronic structure problem is defined according to block spin labeling ). This is opposed to the OpenFermion conventions, that has alternating spin labeling ). When transforming the problem to a Qubit Hamiltonian, described by Pauli strings, different labeling conventions can result in different Hamiltonians, which in turn, might lead to different quantum circuits in terms of depth or cx-counts.
The FermionToQubitMapper class: From Fock space to Qubit space
Transforming to Qubit Hamiltonian (Pauli strings)
Typically, when dealing with Fock space operators we need to transform the creation/annihilation operators to Pauli operators, suitable for quantum algorithms. There are several known transforms, such as Jordan Wigner (JW) and Bravyi Kitaev (BK) transforms.Output:
Hartree Fock state
Once we have a problem and a mapper in hand, we can construct some quantum primitives. One example is the Hartree Fock state, typically used as an initial condition for ground state solvers. For the Jordan Wigner or the Bravyi Kitaev transforms, the Hartree Fock state, which is an elementary basis state in the Fock space, is mapped into a single computational basis state. This state can be determined using theget_hf_state function.
Output:
HF state under the JW transform: : Working with the JW transform, there is a simple relation between the original Fock (occupation number) and transformed (computational) basis states: the state corresponds to occupation of the -th spin orbital in both spaces. Therefore, the Hartree Fock state under this transformation is the string . (This is as opposed to the BK transform, that gives a different computational basis state).
The Z2SymTaperMapper class: From Fock space to reduced Qubit space by using symmetries
Using symmetries of the second quantized Hamiltonian, one can reduce the number of qubits representing the problem by removing (tapering) qubits. The theory of qubit tapering is broad and complex, see for example Refs [3] and [4]. The
Z2SymTaperMapper defines a mapper that includes qubit tapering. It can be initialized by providing symmetries data (set of generators and Pauli operators) explicitly, or by providing a Fermionic Hamiltonian problem. In the latter case, that is introduced below, the symmetries are deduced from the problem Hamiltonian.
This is done via the from_problem method.
**In the following section we provide some mathematical explanation of what is happening behind the secens when defining the Z2SymTaperMapper.
All the logic presented below is incorporated as part of this class.
The advanced reader is encouraged to follow this part, while less experienced readers may choose to skip ahead to the Constructing a VQE section without loss of continuity.**
Reducing the problem size with symmetries (qubit tapering)
The main steps of qubit tapering is as follows (see some technical details in Appendix B at the end of this notebook):- Find generators for a group of operators that commute with the Hamiltonian : for all , .
- Find a unitary transformation that diagonalizes all , such that each generator operates trivially on all qubits except one, e.g., they transform to operators of the form for some qubit number . It can be shown that such unitary can be constructed as , where is operating on a some single qubit .
- Apply the transformation , whose eigenspace will be identical to those of .
- Taper off qubits from the transformed Hamiltonian.
Output:
Output:
Intuition for conserved quantities under the JW transform: In electronic structure problems we have, for example, particle number conservation, spin conservation, and number of particles with fixed spin orientation. The latter corresponds to the two Fermionic operators:
As explained in the previous info box, working with the JW transform gives that Fock basis state are trasformed to the computational basis states. In particular, there is a relation between the orbital number operator and the operators: . We cannot use the transformation of as our symmetry generators, since they correspond to a sum of Pauli strings rather than a single string. However, we can use any function of those, >for example , which, up to a global phase, gives the generators
We can see that is indeed a generator in the example above.Next, we can define a transformation from the generators the the Pauli operators, which means that it block-diagonalizes the Hamiltonian according to symmetry subspaces. This unitary is given by .
Output:
- We shall see that after transformation, the Hamiltonian acts trivially on some of the qubits, with the identity or with the operators found above. Thus, we can reduce it by going to one of the two eigenspaces of these operators, with eigenvalues .
Output:
x_ops. We shall choose a sector, i.e., the or subspace, for each of the subspaces operations.
Which eigenspace to choose?
The answer to this question depends on the problem at hand. If we would like to find the minimal energy of the Hamiltonian, then we shall take the subspace containing the minimal energy.
One possibility is to solve multiple () problems on all sectors. However, another approach is to fix the sector according to the HF state, which is assumed to be in the optimal sector with minimal energy.
This is the default sector defined in Z2SymTaperMapper when initializing with the .from_problem method.
To emphasize the effect of choosing different sectors, we construct tapered operators, each for the subspaces (sectors) , and classically calculate the ground state for each tapered Hamiltonian.
Output:
Constructing a VQE model with Classiq
Next, we use all the classical pre-processing and definitions from the previous sections to build, synthesize, and execute a VQE model. We will take the following steps:
- Defining the transformed and tapered-off Hartree Fock state, which serves as an initial condition for the problem.
- Constructing the transformed and tapered-off UCC ansatz.
- Defining, synthesizing, and executing the full model
Output:
Moving to symmetry subspaces: The Hartree Fock and the UCC operators that are defined below do not necessarily have the same symmetries of the molecular Hamiltonian. Thus, after the block-diagonalization, the resulting operators are not restricted to the symmetries’ subspaces. We take the following approach: we remove terms which do not satisfy the symmetry relation, i.e., commute with symmetry generators. This is done automatically by calling the corresponding functions.
- Hartree Fock in the tapered-off space
Output:
- UCC ansatz
- Running a VQE
prepare_basis_state function) and evolving a parametric UCCSD Hamiltonian with Suzuki Trotter.
Output:
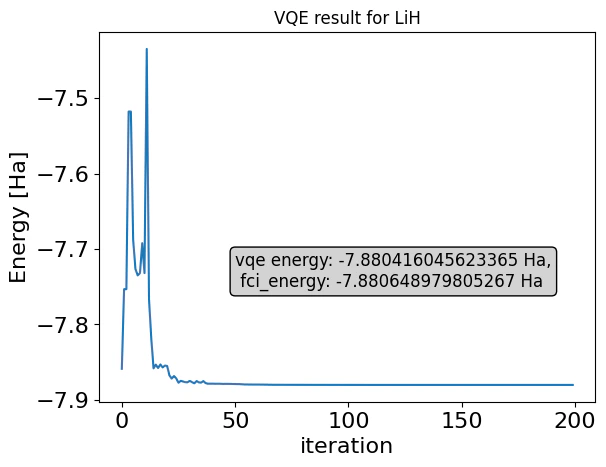
minimize method of ExecutionSession
Output:
Appendix A
- Loading molecule geometry
Appendix B
- Techical details on qubit tapering